
- HOME
- News & Events
- Publications
- 【Publications】Digenic inheritance of mutations in EPHA2 and SLC26A4 in Pendred syndrome
Publications
【Publications】Digenic inheritance of mutations in EPHA2 and SLC26A4 in Pendred syndrome
March 16 2020
Masanori Nakayama
Li M, Nishio S, Naruse C, Riddell M, Sapski S, Katsuno T, Hikita T, Mizapourshafiy F, Smith FM, Cooper LT, Lee MG, Asano M, Boettger T, Krueger M, Wietelmann A, Graumann J, Day BW, Boyd AW, Offermanns S, Kitajiri S, Usami S, Nakayama M*
*Corresponding author
Nature Communications
2020 Mar 12;11(1):1343. doi: 10.1038/s41467-020-15198-9.
Highlights
- Pendred syndrome is a disorder typically associated with hearing loss and thyroid goiter accounting for 7.5% to 15 % of all cases of congenital deafness.
- Approximately 20 years ago, the SLC26A4 gene, encoding pendrin, was reported as a causative gene of this syndrome. While Pendred syndrome is an autosomal recessive disorder, a considerable number of patients carry only mono-allelic mutation on the SCL26A4 gene.
- We identifiy pendrin as a novel binding partner of EphA2, controlling its localization.
- While A subclass ephrins are generally the ligand of EphAs and B subclass ephrins are that of EphB, we show that both ephrin-As and ephrin-B2 are ligand of EphA2.
- We identify EphA2 mutations in patients diagnosed with non-syndromic hearing loss carrying mono-allelic mutation of SLC26A4. Induction of the identified EphA2 mutations reveals a compromised binding ability of EphA2 with ephrin-B2, but not ephrin-A1.
- Our finding uncovers not only an unexpected relationship between the Eph/ephrin system and Pendred syndrome, but also ligand class specific effect on the Eph/eprhin system.
Abstract
Enlarged vestibular aqueducts (EVA) is one of the most commonly identified inner ear malformations in hearing loss patients including Pendred syndrome. While biallelic mutations of the SLC26A4 gene, encoding pendrin, causes non-syndromic hearing loss with EVA or Pendred syndrome, a considerable number of patients appear to carry mono-allelic mutation. This suggests faulty pendrin regulatory machinery results in hearing loss. Here we identify EPHA2 as another causative gene of Pendred syndrome with SLC26A4. EphA2 forms a protein complex with pendrin controlling pendrin localization, which is disrupted in some pathogenic forms of pendrin. Moreover, point mutations leading to amino acid substitution in the EPHA2 gene are identified from patients bearing mono-allelic mutation of SLC26A4. Ephrin-B2 binds to EphA2 triggering internalization with pendrin inducing EphA2 autophosphorylation weakly. The identified EphA2 mutants attenuate ephrin-B2- but not ephrin-A1-induced EphA2 internalization with pendrin. Our results uncover an unexpected role of the Eph/ephrin system in epithelial function.
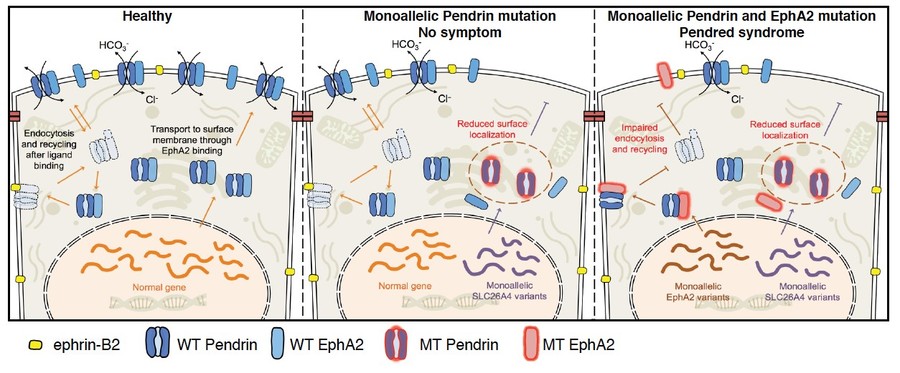
At cell-to-cell contact sites, ephrin-B2 may induce pendrin internalization via EphA2 to remove the protein complex. Mono-allelic mutation on the SLC26A4 gene does not cause the symptom, indicating the single allele of the SLC26A4 gene is sufficient for pendrin function. The pendred syndrome patients carrying the mono-allelic mutation on the SCL26A4 genes with the mono-allelic EPHA2 mutation may cause mislocalization of pendrin at the cell-to-cell contact sites, resulting in reduced functional pendrin on the apical surface of the epithelial cells.
![]()