
- HOME
- News & Events
- Publications
- 【Publications】Systematic analysis of transcription start sites in avian development
Publications
【Publications】Systematic analysis of transcription start sites in avian development
September 7 2017
Guojun Sheng
Paper information
Marina Lizio, Ruslan Deviatiiarov, Hiroki Nagai, Laura Galan, Erik Arner, Masayoshi Itoh, Timo Lassmann, Takeya Kasukawa, Akira Hasegawa, Marian A. Ros, Yoshihide Hayashizaki, Piero Carninci, Alistair R. R. Forrest, Hideya Kawaji*, Oleg Gusev* and Guojun Sheng* (* corresponding authors)
PLOS Biology (doi:10.1371/journal.pbio.2002887)
http://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.2002887
Highlights
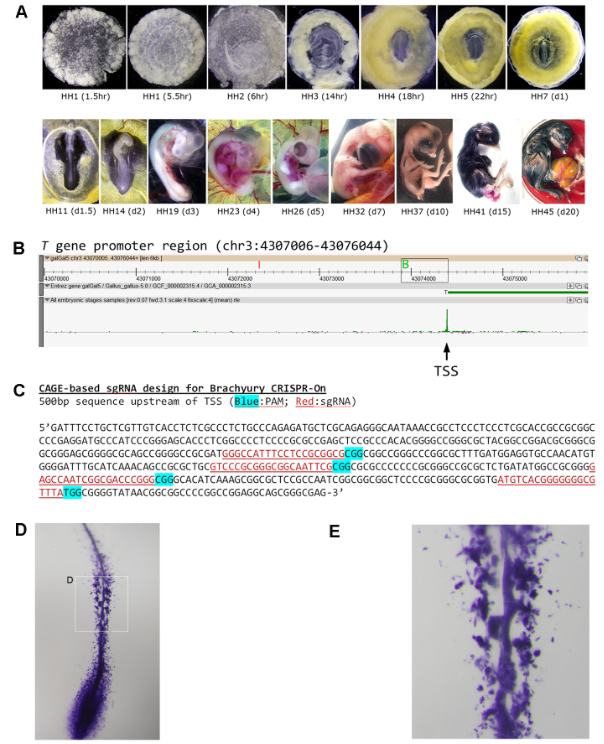
Early development cannot be studied in humans. Analysis of embryogenesis using avian models, phylogenetically closely related to the mammals, can help us understand the complex regulatory mechanism of cell lineage specification in human development. We monitored the three weeks of chicken embryonic development via CAGE expression profiling and revealed the first avian genome-wide set of genuine TSSs critical for differentiation and maturation from pre-gastrulation to hatching. By analyzing stage-specific expression profiles, we have identified enriched transcription factors responsible for lineage commitment and their corresponding regulatory modules. In addition, we reported a set of stable housekeeping genes more suitable for cross-sample normalization and calibration. Finally, we demonstrated the utility of a CAGE-based TSS dataset in developmental studies. Using Brachyury as an example, we showed that CRISPR/Cas9-based genome-editing tools can be efficiently employed to target and transcriptionally activate the promoters of virtually any endogenous genes, enabled by the knowledge of their precise TSS locations. Our data, made available for easy exploration through the open ZENBU platform, represent an invaluable resource to study amniote early development.
Abstract
Cap Analysis of Gene Expression (CAGE) in combination with single-molecule sequencing technology allows precision mapping of transcription start sites (TSSs) and genome-wide capture of promoter activities in differentiated and steady state cell populations. Much less is known about whether TSS profiling can characterize diverse and non-steady state cell populations, such as the approximately 400 transitory and heterogeneous cell types that arise during ontogeny of vertebrate animals. To gain such insight, we used the chick model and performed CAGE-based TSS analysis on embryonic samples covering the full three-week developmental period. In total, 31,863 robust TSS peaks (>1TPM, 1 tag per million) were mapped to the latest chicken genome assembly, of which 34% to 46% were active in any given developmental stage. ZENBU, a web-based open-source platform, was used for interactive data exploration. TSSs of genes critical for lineage differentiation could be precisely mapped and their activities tracked throughout development, suggesting that non-steady states and heterogeneous cell populations are amenable to CAGE-based transcriptional analysis. Our study also uncovered a large set of extremely stable housekeeping TSSs and many novel stage-specific ones. We furthermore demonstrated that TSS mapping could expedite motif-based promoter analysis for regulatory modules associated with stage-specific and housekeeping genes. Finally, using Brachyury as an example, we provide evidence that precise TSS mapping in combination with CRISPR-on technology enables us, for the first time, to efficiently target endogenous avian genes for transcriptional activation. Taken together, our results represent the first report of genome-wide TSS mapping in birds and the first systematic developmental TSS analysis in any amniote species (birds and mammals). By facilitating promoter-based molecular analysis and genetic manipulation, our work also underscores the value of avian models in unravelling the complex regulatory mechanism of cell lineage specification during amniote development.
![]()