Masaya Baba
Paper Information
Hasumi H, Baba M, Hasumi Y, Lang M, Huang Y, Oh HF, Matsuo M, Merino MJ, Yao M, Ito Y, Furuya M, Iribe Y, Kodama T, Southon E, Tessarollo L, Nagashima K, Haines DC, Linehan WM, Schmidt LS. Folliculin-interacting proteins Fnip1 and Fnip2 play critical roles in kidney tumor suppression in cooperation with Flcn. Proc Natl Acad Sci U S A. 2015.
http://www.pnas.org/content/early/2015/03/12/1419502112.long
Highlights
• Flcn is a tumor suppressor, which is responsible for Birt-Hogg-Dubé syndrome, a hereditary kidney cancer syndrome.
• Flcn interacts with homologous proteins Fnip1 and Fnip2, which roles in kidney are unknown.
• Fnip1 knockout mice show identical phenotype to Flcn knockout mice in skeletal muscle and heart, but no phenotype in kidney.
• Fnip2 knockout mice show no phenotype.
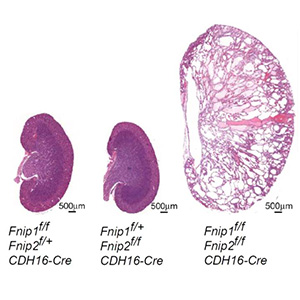
• Kidney-targeted Fnip1/Fnip2 double knockout mice and Fnip1/Fnip2/Flcn triple knockout mice show enlarged polycystic kidneys as a consequence of aberrant cell proliferation, identical to kidney-targeted Flcn knockout mice phenotype.
• Fnip1 heterozygous / Fnip2 homozygous knockout mice develop kidney cancers in 2 years, analogous to Flcn heterozygous knockout mice.
Abstract
Folliculin (FLCN)-interacting proteins 1 and 2 (FNIP1, FNIP2) are homologous binding partners of FLCN, a tumor suppressor for kidney cancer. Recent studies have revealed potential functions for Flcn in kidney; however, kidney-specific functions for Fnip1 and Fnip2 are unknown. Here we demonstrate that Fnip1 and Fnip2 play critical roles in kidney tumor suppression in cooperation with Flcn. We observed no detectable phenotype in Fnip2 knockout mice, whereas Fnip1 deficiency produced phenotypes similar to those seen in Flcn-deficient mice in multiple organs, but not in kidneys. We found that absolute Fnip2 mRNA copy number was low relative to Fnip1 in organs that showed phenotypes under Fnip1 deficiency but was comparable to Fnip1 mRNA copy number in mouse kidney.
Strikingly, kidney-targeted Fnip1/Fnip2 double inactivation produced enlarged polycystic kidneys, as was previously reported in Flcn-deficient kidneys. Kidney-specific Flcn inactivation did not further augment kidney size or cystic histology of Fnip1/Fnip2 double-deficient kidneys, suggesting pathways dysregulated in Flcn-deficient kidneys and Fnip1/Fnip2 double-deficient kidneys are convergent. Heterozygous Fnip1/homozygous Fnip2 double-knockout mice developed kidney cancer at 24 mo of age, analogous to the heterozygous Flcn knockout mouse model, further supporting the concept that Fnip1 and Fnip2 are essential for the tumor-suppressive function of Flcn and that kidney tumorigenesis in human Birt-Hogg-Dubé syndrome may be triggered by loss of interactions among Flcn, Fnip1, and Fnip2. Our findings uncover important roles for Fnip1 and Fnip2 in kidney tumor suppression and may provide molecular targets for the development of novel therapeutics for kidney cancer.